Primeras infecciones mixtas en cerezo por diferentes genotipos del virus de la cereza pequeña (Autora: A.B. Ruiz-García)
27/04/20 14:33 ..... Divulgacion
El virus de la cereza pequeña (Little cherry virus 1 - LChV-1) es un miembro del género Velarivirus de la familia Closteroviridae y tiene importancia en el cultivo del cerezo, ya que se ha asociado a diferentes desórdenes en la planta como enanismo o el síndrome Shirofugen. En el año 2016, nuestro equipo de investigación realizó las primeras detecciones en España de este virus, empleando la tecnología de la secuenciación masiva (high throughput sequencing -HTS), que permitió mediante el análisis de pequeños RNAs interferentes la recuperación completa de la secuencia genómica de dos aislados del virus (KX192366 - aislado Jerte y KX192367 - aislado Ponferrada). Más recientemente en un nuevo estudio, hemos podido recuperar las secuencias completas de dos aislados diferentes (MH300060 - aislado P8-23 y MH300061 - aislado P8-42), que estaban coinfectando un cerezo dulce (cv. Planera) de Alicante, España, con síntomas de enrojecimiento en las hojas.


Este tipo de infecciones mixtas en cerezo con diferentes genotipos de LChV-1 no habían sido descritas hasta la fecha. Ya que no existe vector conocido del virus, la coexistencia de estas variantes podría ser atribuida a prácticas agronómicas como el injerto. Estas infecciones mixtas pueden tener implicaciones en la patogenicidad del virus ya que pueden además producirse recombinaciones. La presencia de dos genotipos diferentes además sugiere dos eventos diferentes de introducción en España.
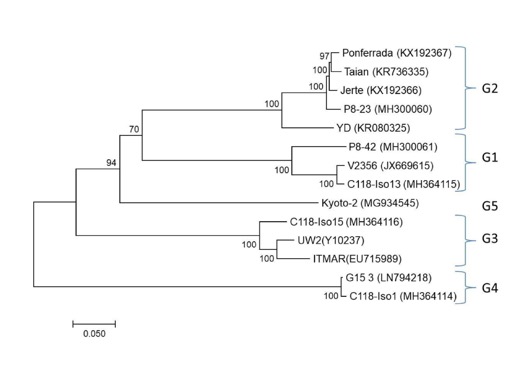
En un estudio que hemos realizado en colaboración con investigadores de Francia, Grecia y Eslovaquia, hemos logrado un avance en el conocimiento de la diversidad del virus ampliando las cuatro agrupaciones o “clusters” que se conocían a cinco. Estos avances permiten el diseño de nuevas herramientas de diagnóstico capaces de detectar todas las variantes de LChV-1 conocidas hasta el momento.
En la figura de abajo se muestra un árbol filogenético (Maximum likelihood; 500 réplicas) que relaciona las secuencias completas disponibles hasta la fecha del LChV-1.
Este tipo de infecciones mixtas en cerezo con diferentes genotipos de LChV-1 no habían sido descritas hasta la fecha. Ya que no existe vector conocido del virus, la coexistencia de estas variantes podría ser atribuida a prácticas agronómicas como el injerto. Estas infecciones mixtas pueden tener implicaciones en la patogenicidad del virus ya que pueden además producirse recombinaciones. La presencia de dos genotipos diferentes además sugiere dos eventos diferentes de introducción en España.
En un estudio que hemos realizado en colaboración con investigadores de Francia, Grecia y Eslovaquia, hemos logrado un avance en el conocimiento de la diversidad del virus ampliando las cuatro agrupaciones o “clusters” que se conocían a cinco. Estos avances permiten el diseño de nuevas herramientas de diagnóstico capaces de detectar todas las variantes de LChV-1 conocidas hasta el momento.
En la figura de abajo se muestra un árbol filogenético (Maximum likelihood; 500 réplicas) que relaciona las secuencias completas disponibles hasta la fecha del LChV-1.
blog comments powered by Disqus
 orcid.org/0000-0001-8406-7963
orcid.org/0000-0001-8406-7963