Primeras detecciones de la falsa sharka en níspero en España (Autora: A.B. Ruiz-García)
03/11/21 11:26 ..... Divulgacion
Continuando con el níspero y avanzar en el conocimiento de su estado sanitario en España, en prospecciones realizadas en áreas productoras de níspero se ha detectado por primera la presencia del virus de la falsa sharka, apple clorotic leaf spot virus (ACLSV) en níspero con una alta incidencia del 31.76%.
La secuencia y caracterización genómica del primer aislado español de ACLSV infectando níspero se realizó de la siguiente forma.
El análisis por secuenciación masiva (HTS) realizado en el ARN total extraído de la muestra SL73.6 rindió 31.867.726 lecturas d con un tamaño promedio de 134,8 nt. Se realizó la sustracción del genoma del níspero, tras el control de calidad de las lecturas y eliminar los adaptadores, lo que resultó en 1.698.300 lecturas que se utilizaron para el ensamblado de novo que dio lugar a 13.797 contigs. Se anotaron los contigs relacionados con virus
y viroides mediante BLASTN/X. Este análisis mostró 11 contigs de tamaños entre
6511 y 281 nt relacionados con ACLSV, confirmando la presencia de ACLSV en la muestra. La extensión de los contigs realizando el mapeo de las lecturas contra los contigs permitió la recuperación de un genoma casi completo de 7533 nt, cubierto por 44 039 lecturas (cobertura promedio 857,2x), aislado llamado SL73.6 y depositado en GenBank, número de acceso OK272502. Este aislado mostró el mayor porcentaje de identidad a nivel nueclotídico (83,7%) con el aislado alemán 38/85-B (KX579123) de manzano. El mapa de la organización del genoma SL73.6, la cobertura promedio de HTS y la similitud de proteínas de sus ORF con el aislado de ACLSV más cercano (KX579123) se muestran en la Figura1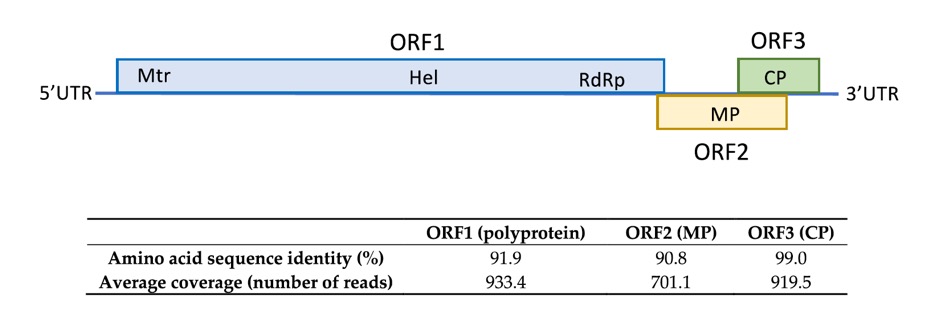
Figura 1. Similaridad aminoacídica del aislado SL73.6 con los ORFs del aislado de ACLSV más próximo (KX579123). Se muestra la cobertura por HTS.
En la siguiente figura se muestra la relación filogenética del aislado de ACLSV de níspero con otros aislados de ACLSV de otros orígenes y de otros hospedados.

La secuencia y caracterización genómica del primer aislado español de ACLSV infectando níspero se realizó de la siguiente forma.
El análisis por secuenciación masiva (HTS) realizado en el ARN total extraído de la muestra SL73.6 rindió 31.867.726 lecturas d con un tamaño promedio de 134,8 nt. Se realizó la sustracción del genoma del níspero, tras el control de calidad de las lecturas y eliminar los adaptadores, lo que resultó en 1.698.300 lecturas que se utilizaron para el ensamblado de novo que dio lugar a 13.797 contigs. Se anotaron los contigs relacionados con virus
y viroides mediante BLASTN/X. Este análisis mostró 11 contigs de tamaños entre
6511 y 281 nt relacionados con ACLSV, confirmando la presencia de ACLSV en la muestra. La extensión de los contigs realizando el mapeo de las lecturas contra los contigs permitió la recuperación de un genoma casi completo de 7533 nt, cubierto por 44 039 lecturas (cobertura promedio 857,2x), aislado llamado SL73.6 y depositado en GenBank, número de acceso OK272502. Este aislado mostró el mayor porcentaje de identidad a nivel nueclotídico (83,7%) con el aislado alemán 38/85-B (KX579123) de manzano. El mapa de la organización del genoma SL73.6, la cobertura promedio de HTS y la similitud de proteínas de sus ORF con el aislado de ACLSV más cercano (KX579123) se muestran en la Figura1
Figura 1. Similaridad aminoacídica del aislado SL73.6 con los ORFs del aislado de ACLSV más próximo (KX579123). Se muestra la cobertura por HTS.
En la siguiente figura se muestra la relación filogenética del aislado de ACLSV de níspero con otros aislados de ACLSV de otros orígenes y de otros hospedados.
blog comments powered by Disqus
 orcid.org/0000-0001-8406-7963
orcid.org/0000-0001-8406-7963