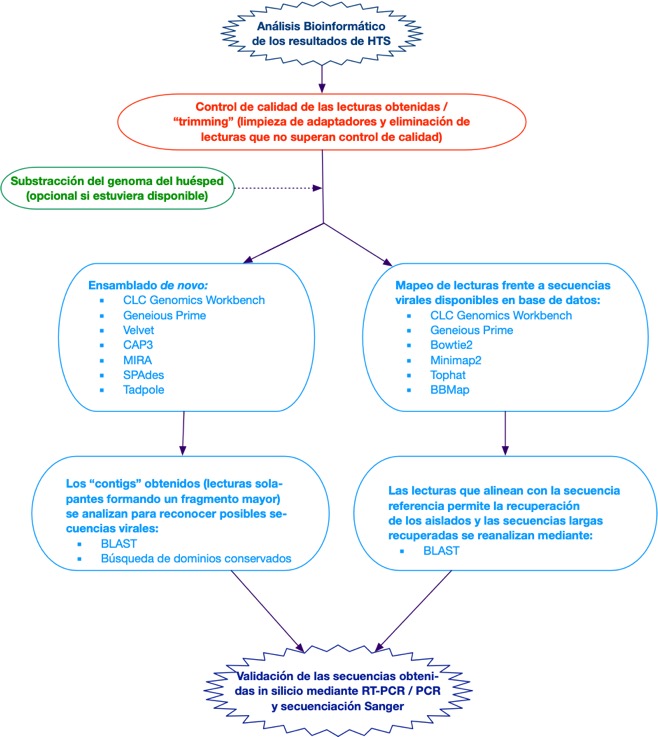
¿Cuál es el esquema del análisis bioinformático de los resultados de secuenciación masiva para el estudio de virus? (Autora: A.B. Ruiz-García)
Los viroides ¿qué son? (Autora: A.B. Ruiz-García)
Su genoma es una molécula de RNA monocatenario circular de 270 a 470 nucleótidos, que no está encapsidada y no es codificante. No tienen pues proteínas propias y son parásitos obligados de las células de la planta huésped.
Se replican de forma autónoma utilizando el sistema de transcripción de las células infectadas. Siguen el modelo de realización del círculo rodante y está relacionado con la naturaleza circular de los viroides.
Hay dos familias de viroides:
Pospiviroidae que se replican en el núcleo
Avsunviroidae que se replican en los cloroplastos
Los viroides invaden la planta huésped por una herida inicial y movimiento célula a célula via plasmodesmos e invasión a larga distancia por los tejidos conductores del floema
La transmisión planta a planta es:
- principalmente mecánica
- pocos casos por pulgones, cuando una molécula de viroide se encapsida en partículas de virus en confección virus-viroide
- por multiplicación vegetativa
- semilla
Los síntomas son similares a los virus fitopatógenos, raquitismo, epinastia, decoloraciones foliares, aclarado de nervios, necrosis, distorsión de hojas, lesiones necróticas locales o generalizadas
El primer viroide descubierto fue el causante de la enfermedad del tubérculo fusiforme de la patata Potato splindle tuber viroid o PSTVd
El segundo viroide fue el de la exocortis de los cítricos, citrus exocortis viroid o CEVd

otro viroide de importancia es el Peach latente mosaic viroid o PLMVd que tiene una sintomatología muy característica en los frutos
Clasificación de virus vegetales de DNA de cadena simple (Autora: A.B. Ruiz-García)
Los más representativos se clasifican en los siguientes géneros
Familia Geminiviridae
Géneros:
* Becurtovirus
* Begomovirus
* Capulavirus
* Curtovirus
* Grablovirus
* Eragrovirus
* Mastrevirus
* Topocuvirus
* Turncurtovirus
Familia Nanoviridae
Géneros:
* Babuvirus
* Nanovirus
¿Cómo evitar la introducción de cepas agresivas tipo M de sharka? (Autora: A.B. Ruiz-García)
temprana de melocotonero en amplias zonas de un país como España que
produce más de 700.000 toneladas de melocotones.
Hay unas medidas importantes que deben conocerse y aplicarse para evitar o reducir los riesgos de
introducción de tipos agresivos del virus de la sharka:
- La más básica: No introducir material vegetal de zonas donde exista PPV M, evitando la toma de varetas de material vegetal especialmente si son recolectadas en el sur de Francia, Italia, Grecia o cualquier país de Europa del Este.
- Una medida lógica: No plantar material vegetal de frutales de hueso sin un análisis previo de PPV y que las plantaciones se realicen con material certificado libre de PPV
- Una medida de atención: Vigilar los árboles en floración, durante el engrosamiento del fruto y en el momento de la recolección y observar si aparecieran síntomas
- Una medida más: Analizar los melocotoneros que presenten síntomas de PPV
¿Por qué no es suficiente con la sensibilidad y la especificidad en el diagnóstico? (Autora: A.B. Ruiz-García)
Fijémonos en la tabla de contingencia de variables de diagnóstico:
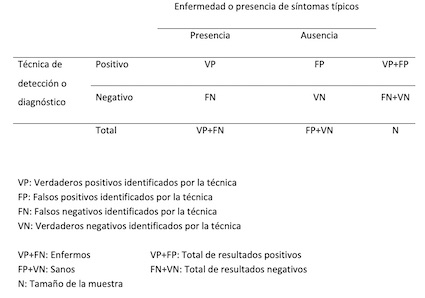
La sensibilidad de la prueba diagnóstica se define como la probabilidad de que el resultado de la prueba sea positivo en una planta enferma. Representa la proporción de verdaderos positivos diagnosticados por la técnica entre las plantas enfermas VP/(VP+FN)
La especificidad de la prueba diagnóstica se define como la probabilidad de que el resultado de la prueba sea negativo en una planta sana. Representa el porcentaje de resultados negativos respecto del total de plantas sanas VN/(FP+VN)
La sensibilidad no contempla los falsos positivos y la especificidad no contempla los falsos negativos que rinde la técnica. Y es precisamente esto lo que necesita conocer el técnico o lo que es lo mismo saber ¿cúal es la probabilidad de que un resultado positivo por la técnica sea un verdadero positivo y no un falso positivo, independientemente de su sensibilidad? y ¿cúal es la probabilidad de que un resultado negativo por la técnica sea un verdadero negativo y no un falso negativo, independientemente de su especificidad? La respuesta la dan los valores predictivos positivo y negativo, que se calculan considerando las filas de la tabla de contingencia al contrario que la sensibilidad y especificidad que se calculan en las columnas de la tabla de contingencia.
El valor predictivo positivo de una prueba diagnóstica se define como la probabilidad de que la planta esté enferma habiendo dado positivo en la prueba. Así pues, el valor predictivo positivo representa el porcentaje de plantas realmente enfermas respecto del total de plantas que han dado positivo VP/(VP+FP). Un valor alto indica que la probabilidad de que la planta esté realmente enferma habiendo dado positivo en la prueba diagnóstica es muy alta.
El valor predictivo negativo se define como la probabilidad de que una planta esté sana habiendo dado negativo. Representa el porcentaje de plantas sanas respecto del total de plantas que han dado negativo en la prueba VN/(VN+FN).
Sin embargo, si aplicamos el teorema de Bayes se puede concluir que los valores predictivos dependen de la prevalencia de la enfermedad que tratamos de diagnosticar según la siguiente relación: valor predictivo positivo= (sensibilidad de la prueba x prevalencia de la enfermedad) / ((sensibilidad x prevalencia) + [(1 - especificidad) x (1 - prevalencia)]). Como puede observarse, cuanto mayor sea la prevalencia de la enfermedad en la población mayor será el valor predictivo positivo de la prueba diagnóstica, aún manteniéndose constantes su sensibilidad y especificidad; de forma similar ocurre con el valor productivo negativo.
Así pues, si sensibilidad y especificidad no dan una respuesta apropiada por no contemplar falsos positivos y falsos negativos respectivamente y los valores predictivos positivo y negativo varían con la prevalencia ¿qué parámetros son los apropiados para evaluar la técnica diagnóstica? La respuesta la dan “likelihood ratios” de positivo y negativo o razón de verosimilitud. Así la razón de verosimilitud positiva es la proporción de verdaderos positivos correctamente identificados por la técnica, o sea, la sensibilidad dividido por la proporción de falsos positivos que la técnica diagnostica FP/VP+FP, o lo que es lo mismo 1-especificidad = 1-(VN/(VN+FP)).
La razón de verosimilitud negativa es la proporción de falsos negativos que la técnica diagnostica (1-sensibilidad), dividido por la proporción de verdaderos negativos correctamente identificados por la técnica o lo que es lo mismo, la especificidad
La enorme ventaja de emplear las razones de verosimilitud es que pueden usarse para cuantificar la probabilidad de la enfermedad para una planta individual. El teorema de Bayes se usa para traducir la información suministrada por los las razones de verosimilitud y probabilidad de enfermedad. El teorema de Bayes establece que la razón de probabilidades o razón de momios pre-test, que es lo mismo el cociente de la probabilidad de enfermedad y la probabilidad de no enfermedad antes de realizar el test, multiplicado por la razón de verosimilitud da lugar a la razón de probabilidades post-test de la enfermedad o sea el cociente de la probabilidad de la enfermedad y la probabilidad de no enfermedad después de realizar el análisis. La probabilidad post-test se puede calcular de la siguiente forma:
Probabilidad Pre-test = Prevalencia
Razón de probabilidades Pre-test = Prevalencia/(1-Prevalencia)
Razón de probabilidades Post-test = Razón de probabilidades Pre-test x Razón de verosimilitud
Probabilidad Post-test = Razón de probabilidades Post-test /(1+ Razón de probabilidades Post-test)
Además las razones de verosimilitud de diversos métodos pueden combinarse en la formula de modo que: Razón de probabilidades Post-test = Razón de probabilidades Pre-test x Razón de verosimilitud 1 x Razón de verosimilitud 2 x Razón de verosmilitud 3.
Así pues la probabilidad pre-test de enfermedad puede compararse con la probabilidad post-test. La diferencia entre la previa y la posterior es una manera muy efectiva de analizar la eficiencia de un método diagnóstico.
El enanismo de la satsuma (Autora: A.B. Ruiz-García)

El virus se transmite rápido por injerto entre cítricos y mecánicamente entre cítricos y huéspedes no cítricos. Su dispersión natural parece ser que es por vectores del suelo en las comarcas de Japón y también se ha indicado una posible transmisión entre Viburnum odoratissimum y cítricos.
La enfermedad produce raquitismo y las hojas se abarquillan y adquieren forma de cuchara.
Los frutos son de baja calidad y bajo valor comercial y es una amenaza para los países productores.
El enanismo de la satsuma severa es un problema en Japón.
Amenazas del exterior II: otro virus de vid peligroso (Autora: A.B. Ruiz-García)
Es importante conocer cómo poder detectarlo en material vegetal por PCR, por lo que abajo hay un protocolo de PCR para su diagnóstico.
Muestreo: 4 brotes terminales (10 cm) alrededor de la cepa
Purificación de ácidos nucleicos: DNeasy Plant kit o similar
Preparación del cóctel: Para cada reacción de 25 microlitros (3 microlitros de DNA purificado)
H2O 10,8 microlitros
Tampón 5x Promega 5 microlitros
Cl2Mg 25 mM (uso 1,5 mM) 1,5 microlitros
dNTP (2,5 mM cada uno) uso 0,25 mM 2,5 microlitros
GVCV-F1 (25 microM) uso 1microM 1 microlitro
GVCV-R1 (25 microM) uso 1microM 1 microlitro
GoTaq G2 Promega (5 U/microlitros) 0,2 microlitros
ORF1 (557pb)
GVGF1 5′-CTCGTCGCATTTGTAAGA-3′ (Al Rwahnih et al.)
GVGR1 5′-ACTGACAAGGCCTACTACG-3′ (Al Rwahnih et al.)
Condiciones del Termociclador
4 min ------------------- 94ºC
40 ciclos: 30 s ---------------------- 92ºC
30 s ---------------------- 50ºC
1 min -------------------- 72ºC
10 min ----------------- 72ºC
Amenazas del exterior I: un virus de vid peligroso (Autora: A.B. Ruiz-García)
Entre ellos está un Badnavirus, un virus de DNA, que se denomina grapevine vein clearing virus. Ha sido el primer virus de DNA descrito en vid (familia Caulimoviridae, género Badnavirus), que produce el síndrome de decaimiento y aclaramiento de las nervaduras y que ha sido descrito en los estados de Missouri, Illinois e Indiana (EEUU).
El protocolo para su detección por PCR convencional es el siguiente:
Muestreo: 4 brotes terminales (10 cm) alrededor de la cepa
Purificación de ácidos nucleicos: DNeasy Plant kit o similar
Preparación del cóctel: Para cada reacción de 25 microlitros (3 microlitros de DNA purificado)
H2O 10,8 microlitros
Tampón 5x Promega 5 microlitros
Cl2Mg 25 mM (uso 1,5 mM) 1,5 microlitros
dNTP (2,5 mM cada uno) uso 0,25 mM 2,5 microlitros
GVCV-F1 (25 microM) uso 1microM 1 microlitro
GVCV-R1 (25 microM) uso 1microM 1 microlitros
GoTaq G2 Promega (5 U/microlitro) 0,2 microlitros
ORF3 (530pb)
GVCV-F1 5′-CACGTTTCAAAGAAAGATGGAC-3′ (Zhang et al.)
GVCV-R1 5′-ATCCKTCCATGCAWCCGTCAG-3′ (Zhang et al.)
Condiciones del Termociclador
4 min ------------------- 94ºC
40 ciclos: 30 s ---------------------- 92ºC
30 s ---------------------- 50ºC
1 min -------------------- 72ºC
10 min ----------------- 72ºC
Metodología bayesiana con modelos latentes (Autora: A.B. Ruiz-García)
A continuación se detalla la programación del modelo:
Código de OpenBUGS, que asume dos técnicas condicionalmente dependientes DAS-ELISA (ELISA) y spot real-time RT-PCR (spot), y una tercera independiente, real-time RT-PCR convencional (qPCR), y tres poblaciones. Priors informativos para sensibilidad (Se) y especificidad (Sp) de DAS-ELISA y real-time RT-PCR convencional. Priors no informativos, β (1,1), para sensibilidad y especificidad de spot real-time RT-PCR y prevalencias de tres poblaciones (Prev1, Prev2, Prev3). Likelihood ratios (LR) se calculan en cada iteración.
model;
{
y1[1:Q, 1:Q, 1:Q] ~ dmulti(p1[1:Q, 1:Q, 1:Q], n1)
y2[1:Q, 1:Q, 1:Q] ~ dmulti(p2[1:Q, 1:Q, 1:Q], n2)
y3[1:Q, 1:Q, 1:Q] ~ dmulti(p3[1:Q, 1:Q, 1:Q], n3)
p1[1,1,1] <- Prev1*SeqPCR*(Sespot*SeELISA+covDp) + (1-Prev1)*(1-SpqPCR)*((1-Spspot)*(1-SpELISA)+covDn)
p1[1,2,1] <- Prev1*SeqPCR*(Sespot*(1-SeELISA)-covDp) + (1-Prev1)*(1-SpqPCR)*((1-Spspot)*SpELISA-covDn)
p1[1,1,2] <- Prev1*(1-SeqPCR)*(Sespot*SeELISA+covDp) + (1-Prev1)*SpqPCR*((1-Spspot)*(1-SpELISA)+covDn)
p1[1,2,2] <- Prev1*(1-SeqPCR)*(Sespot*(1-SeELISA)-covDp) + (1-Prev1)*SpqPCR*((1-Spspot)*SpELISA-covDn)
p1[2,1,1] <- Prev1*SeqPCR*((1-Sespot)*SeELISA-covDp) + (1-Prev1)*(1-SpqPCR)*(Spspot*(1-SpELISA)-covDn)
p1[2,2,1] <- Prev1*SeqPCR*((1-Sespot)*(1-SeELISA)+covDp) + (1-Prev1)*(1-SpqPCR)*(Spspot*SpELISA+covDn)
p1[2,1,2] <- Prev1*(1-SeqPCR)*((1-Sespot)*SeELISA-covDp) + (1-Prev1)*SpqPCR*(Spspot*(1-SpELISA)-covDn)
p1[2,2,2] <- Prev1*(1-SeqPCR)*((1-Sespot)*(1-SeELISA)+covDp) + (1-Prev1)*SpqPCR*(Spspot*SpELISA+covDn)
p2[1,1,1] <- Prev2*SeqPCR*(Sespot*SeELISA+covDp) + (1-Prev2)*(1-SpqPCR)*((1-Spspot)*(1-SpELISA)+covDn)
p2[1,2,1] <- Prev2*SeqPCR*(Sespot*(1-SeELISA)-covDp) + (1-Prev2)*(1-SpqPCR)*((1-Spspot)*SpELISA-covDn)
p2[1,1,2] <- Prev2*(1-SeqPCR)*(Sespot*SeELISA+covDp) + (1-Prev2)*SpqPCR*((1-Spspot)*(1-SpELISA)+covDn)
p2[1,2,2] <- Prev2*(1-SeqPCR)*(Sespot*(1-SeELISA)-covDp) + (1-Prev2)*SpqPCR*((1-Spspot)*SpELISA-covDn)
p2[2,1,1] <- Prev2*SeqPCR*((1-Sespot)*SeELISA-covDp) + (1-Prev2)*(1-SpqPCR)*(Spspot*(1-SpELISA)-covDn)
p2[2,2,1] <- Prev2*SeqPCR*((1-Sespot)*(1-SeELISA)+covDp) + (1-Prev2)*(1-SpqPCR)*(Spspot*SpELISA+covDn)
p2[2,1,2] <- Prev2*(1-SeqPCR)*((1-Sespot)*SeELISA-covDp) + (1-Prev2)*SpqPCR*(Spspot*(1-SpELISA)-covDn)
p2[2,2,2] <- Prev2*(1-SeqPCR)*((1-Sespot)*(1-SeELISA)+covDp) + (1-Prev2)*SpqPCR*(Spspot*SpELISA+covDn)
p3[1,1,1] <- Prev3*SeqPCR*(Sespot*SeELISA+covDp) + (1-Prev3)*(1-SpqPCR)*((1-Spspot)*(1-SpELISA)+covDn)
p3[1,2,1] <- Prev3*SeqPCR*(Sespot*(1-SeELISA)-covDp) + (1-Prev3)*(1-SpqPCR)*((1-Spspot)*SpELISA-covDn)
p3[1,1,2] <- Prev3*(1-SeqPCR)*(Sespot*SeELISA+covDp) + (1-Prev3)*SpqPCR*((1-Spspot)*(1-SpELISA)+covDn)
p3[1,2,2] <- Prev3*(1-SeqPCR)*(Sespot*(1-SeELISA)-covDp) + (1-Prev3)*SpqPCR*((1-Spspot)*SpELISA-covDn)
p3[2,1,1] <- Prev3*SeqPCR*((1-Sespot)*SeELISA-covDp) + (1-Prev3)*(1-SpqPCR)*(Spspot*(1-SpELISA)-covDn)
p3[2,2,1] <- Prev3*SeqPCR*((1-Sespot)*(1-SeELISA)+covDp) + (1-Prev3)*(1-SpqPCR)*(Spspot*SpELISA+covDn)
p3[2,1,2] <- Prev3*(1-SeqPCR)*((1-Sespot)*SeELISA-covDp) + (1-Prev3)*SpqPCR*(Spspot*(1-SpELISA)-covDn)
p3[2,2,2] <- Prev3*(1-SeqPCR)*((1-Sespot)*(1-SeELISA)+covDp) + (1-Prev3)*SpqPCR*(Spspot*SpELISA+covDn)
ls <- (Sespot-1)*(1-SeELISA)
us <- min(Sespot,SeELISA) - Sespot*SeELISA
lc <- (Spspot-1)*(1-SpELISA)
uc <- min(Spspot,SpELISA) - Spspot*SpELISA
rhoD <- covDp / sqrt(Sespot*(1-Sespot)*SeELISA*(1-SeELISA))
rhoDc <- covDn / sqrt(Spspot*(1-Spspot)*SpELISA*(1-SpELISA))
covDn ~ dunif(lc, uc)
covDp ~ dunif(ls, us)
SeELISA~dbeta(108.41,18.49)
SpELISA~dbeta(34.17,1.34)
Sespot~dbeta(1,1)
Spspot~dbeta(1,1)
SeqPCR~dbeta(53.58,2.63)
SpqPCR~dbeta(51.41,7.23)
Prev1~dbeta(1,1)
Prev2~dbeta(1,1)
Prev3~dbeta(1,1)
LRELISApos<-SeELISA/(1-SpELISA)
LRELISAneg<-(1-SeELISA)/SpELISA
LRspotpos<-Sespot/(1-Spspot)
LRspotneg<-(1-Sespot)/Spspot
LRqPCRpos<-SeqPCR/(1-SpqPCR)
LRqPCRneg<-(1-SeqPCR)/SpqPCR
LRELISAposspotpos<-LRELISApos*LRspotpos
LRELISAposspotneg<-LRELISApos*LRspotneg
LRELISAnegspotpos<-LRELISAneg*LRspotpos
LRELISAnegspotneg<-LRELISAneg*LRspotneg
LRELISAposqPCRpos<-LRELISApos*LRqPCRpos
LRELISAposqPCRneg<-LRELISApos*LRqPCRneg
LRELISAnegqPCRpos<-LRELISAneg*LRqPCRpos
LRELISAnegqPCRneg<-LRELISAneg*LRqPCRneg
LRspotposqPCRpos<-LRspotpos*LRqPCRpos
LRspotposqPCRneg<-LRspotpos*LRqPCRneg
LRspotnegqPCRpos<-LRspotneg*LRqPCRpos
LRspotnegqPCRneg<-LRspotneg*LRqPCRneg
LRELISAposspotposqPCRpos<-LRELISApos*LRspotpos*LRqPCRpos
LRELISAposspotposqPCRneg<-LRELISApos*LRspotpos*LRqPCRneg
LRELISAposspotnegqPCRpos<-LRELISApos*LRspotneg*LRqPCRpos
LRELISAposspotnegqPCRneg<-LRELISApos*LRspotneg*LRqPCRneg
LRELISAnegspotposqPCRpos<-LRELISAneg*LRspotpos*LRqPCRpos
LRELISAnegspotposqPCRneg<-LRELISAneg*LRspotpos*LRqPCRneg
LRELISAnegspotnegqPCRpos<-LRELISAneg*LRspotneg*LRqPCRpos
LRELISAnegspotnegqPCRneg<-LRELISAneg*LRspotneg*LRqPCRneg
}
#Note: for this model initial values were:
#SeELISA=0.86,SpELISA=0.99,SeqPCR=0.97,SpqPCR=0.89
Con estos parámetros se puede establecer la probabilidad post-test de que una cepa esté infectada según los resultados del diagnóstico de una, dos o tres técnicas. De esta forma dependiendo de la prevalencia de la infección se puede seleccionar el método más apropiado para realizar el análisis.
¿Cómo se interpreta una curva de probabilidad post-test en diagnóstico? (Autora: A.B. Ruiz-García)
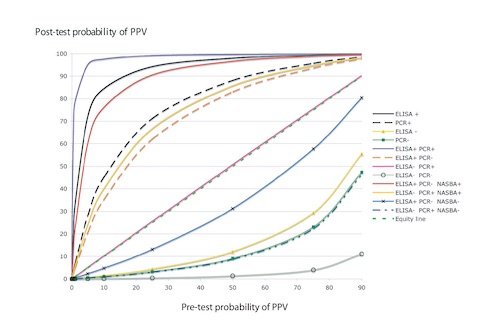
La probabilidad pre-test es la prevalencia de la enfermedad en el área de estudio o en el conjunto de muestras analizadas. la linea recta diagonal divide los resultados positivos (arriba de ella) de los negativos (debajo de ella).
Así como se puede observar a baja prevalencia el ELISA+ junto con una PCR+ dan una probabilidad post-test, es decir una seguridad de que la muestra sea positiva muy alta. Comparando un resultado positivo en este modelo por ELISA y por PCR vemos que a baja prevalencia un resultado positivo es más fiable que un resultado positivo por PCR que tiene una probabilidad post-test más bajo. A altas prevalencia los resultados positivos tienden a igualarse en su probabilidad.
¿Pero y que pasa para los resultados negativos? A bajas prevalencias tanto ELISA- como PCR- como ELISA y PCR- tienden a ser similares sus probabilidades post test, luego cualquiera tiene bastante fiabilidad a bajas prevalecías para los resultados negativos. A altas prevalencia es mejor la PCR porque su probabilidad post-test es decir un falso negativo es menor que el ELISA que un negativo es posible que sea un falso negativo.
Con este tipo de curvas es posible aplicar técnicas de diagnóstico con criterio.
Protocolo IPPC/FAO DP2 Plum pox virus (Autora: A.B. Ruiz-García)
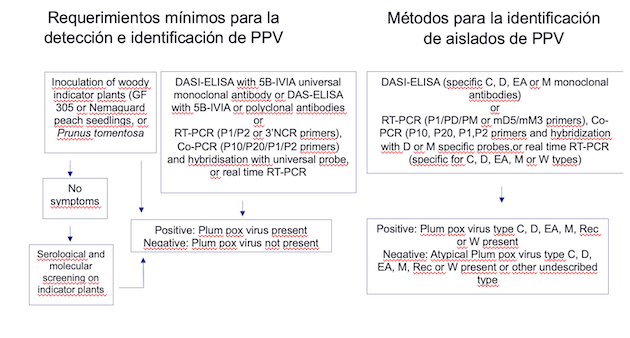
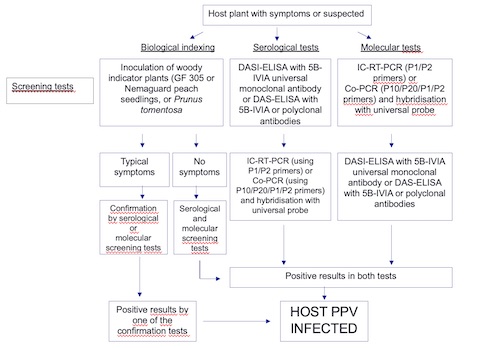
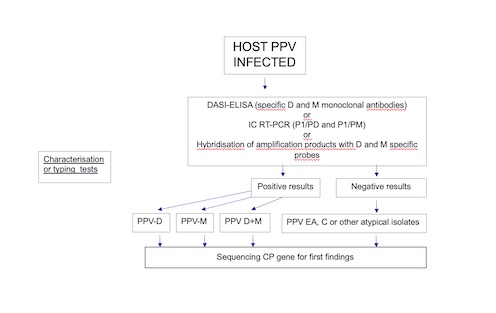
El protocolo oficial de la EPPO para la detección del virus de la sharka (Autora: A.B. Ruiz-García)
El enanismo clorótico de los cítricos (Autora: A.B. Ruiz-García)
El rendimiento y producción de los árboles afectados se reduce sustancialmente.
El agente causal el Citrus cholortic dwarf associated virus que tiene como huéspedes, limonero, pomelo, mandarino tangelo y naranjo dulce.

Se transmite por mosca blanca

En campo afecta a todas las variedades de limonero y la mayor parte de mandarinos e híbridos
Sus síntomas son moderados en pomelo y suaves en naranjo dulce lo que puede suponer que al no ser severos sirvan como reservorios del virus y fuente de inóculo.
El tamaño de la planta se reduce las hojas se deforman y hay una fuerte clorosis.
 orcid.org/0000-0001-8406-7963
orcid.org/0000-0001-8406-7963